韦翰斯骨骼系统遗传病检测panel包含300多个与骨骼系统遗传病相关的基因,选自各大权威数据库(OMIM,HPO&CHPO,GeneCard等),最新的文献以及临床大样本数据,为临床疾病的诊断提供全面的指导。
人体骨骼是一个复杂的器官,有206块不同形状和大小的骨组成,具有多种胚胎起源和发挥多方面的功能,如机械支持,保护内脏器官,血液和矿物质储存等。在已经发现的单基因遗传病中,有500多种疾病累及骨骼系统,涉及的致病基因达300多个。在新生儿中的发病率达到1/3,000-1/5,000。包括软骨发育异常,骨密度/强度异常,颅面部发育异常,躯干骨发育异常,四肢骨发育异常和其他类型骨骼发育异常。
成骨不全症(osteogenesis imperfectas,OI)[OMIM 166200]又称脆骨病。是一组以骨骼脆性增加及胶原代谢紊乱为特征的全身性结缔组织疾病。群体发病率约为1/15 000,而中国人群中的发病率约为0.04%。其病变不仅限于骨骼,还常常累及其他结缔组织如眼、耳、皮肤、牙齿等,其特点是骨脆性增加、骨关节进行性畸形、蓝巩膜、牙本质发育不全、听力下降及皮肤异常。
目前至少有4种不同的分子机制导致OI发病:(1) I型前胶原COL1A1和COL1A2基因突变导致胶原合成量减少或结构异常,约占OI患者的90%;(2)CRTAP,P3H1,CyPB共同组成的脯氨酞3-羟化复合体的缺陷造成I型前胶原翻译后被过度修饰;(3)胶原分子伴侣FKBP65和HSP47活性缺失及BMP1缺陷引起I型胶原加工、装配和分泌异常;(4)转录因子OSX突变影响成骨细胞正常分化和SERPINFI缺陷造成骨代谢紊乱.
1.Pagon R A, Bird T D, Dolan C R, et al. COL1A1/2-Related Osteogenesis Imperfecta[J]. 2005.
2.Shapiro J R, Lietman C, Grover M, et al. Phenotypic variability of osteogenesis imperfecta type V caused by an IFITM5 mutation[J]. Journal of Bone and Mineral Research, 2013, 28(7): 1523-1530.
3.石长贵, 张颖, 袁文. 成骨不全治疗研究进展[J]. 脊柱外科杂志, 2013 (3): 178-181.
临床表现典型,疑似骨骼系统病患者,为明确病因进行检测;
临床表现复杂,对应疾病模糊,仅依据某些具体临床体征,难以确诊的患者做鉴别诊断。
对已经生育一胎骨骼系统疾病的夫妇进行检测,评估再次生育患儿的风险。
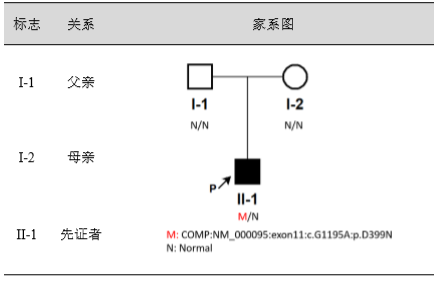
临床信息:男,11岁,双侧髋关节及膝关节正位片提示骨骺发育不良,生长发育迟缓,骶尾骨有隐性脊柱裂。出生正常,三岁后出现乏力,走路不稳。临床诊断骨骺发育不良。
检测内容:韦翰斯骨骼系统遗传病检测panel
检测结果:发现先证者COMP基因上的1个杂合错义变异 c.G1195A(p.D399N),父母均未检出该变异,推测为新发变异,或父母一方存在生殖细胞嵌合。COMP基因异常导致常染色体显性遗传的多发性骨骺发育不良1型(EPIPHYSEAL DYSPLASIA, MULTIPLE, 1; EDM1; OMIM#132400)和假性软骨发育不良(PSEUDOACHONDROPLASIA; PSACH; OMIM#177170)。

先证者COMP 基因中,发现一个杂合变异c.G1195A(p.D399N),该变异导致COMP 基因的第399位密码子由编码天冬氨酸变为天冬酰胺。一代测序结果显示,先证者父母均未检出该变异,推测为新发变异,或父母一方存在生殖细胞嵌合,家系图如下图所示: